研究热休克蛋白90(Hsp90)的MEEVD C-末端肽与热休克调节蛋白(Hop)的四重螺旋重复A(TPR2A)结构域之间的结合是研究Hop-Hsp90相互作用的基本分子细节的有用原型。这篇文章在这里研究了MEEVD肽与TPR2A结构域的结合/解离机制,并使用自适应偏置力(ABF)方法计算了MEEVD肽与TPR2A结构域的标准结合自由能和平均力势。我们观察到结合引发了肽和蛋白质受体的构象变化。文章中使用MMPBSA方法计算测量到了-8.4 kcal/mol的结合自由能,与其在实验中的估计值一致。模拟过程中出现了多次解离和重新结合事件,沿着连接结合位点和溶剂的一致路径进行。MEEVD肽首先通过破坏氢键逐渐解离,然后倾斜在一侧,同时保持与肽的Asp5残基侧链的氢键相互作用。在这种初始位移之后,肽完全解离并进入溶剂中。MEEVD肽从溶剂重新结合到受体结合位点的过程通过入口门缓慢进行。解离和重新结合经历了中间状态,其中肽与受体的一个侧向螺旋helix A1发生相互作用,主要涉及肽的Asp5,Val4和Glu3。这种新发现的中间结构与受体发生了多个接触,进一步促使了蛋白质多肽复合物的形成。重新结合后得到的结合复合物的结构与晶体结构非常相似,其前后的RMSD差距不超过0.48 A。Asp 5,Val 4和Glu 3残基的构象和分子间接触与天然构象具有出色的结构相似性。最后,MEEVD的解离和重新关联导致了水合/脱水转变,在接下里的内容中,我将会为大家较为详细的阐述有关ABF增强采样方法,US伞形采样以及GaMD(加速分子动力学),这三种主要可以用于研究多肽解离机制的方法与在各大软件中的具体实现方法(Amber,GROMACS为主)。
2.ABF enhanced Sampling
现如今增强采样的方法有很多,包括但不限于metadynmics,(e)ABF(自适应偏置力方法),GaMD(高斯加速动力学,集成于Amber中),REUS,SMD(拉伸动力学)等,每一种方法都存在各自的缺陷与优势,下面将主要围绕ABF的具体实现与原理来阐述其与其他的方法之间的主要区别。在使用这一些重要性采样方法的时候需要为体系确立合适的反应坐标,根据目前自己个人的经验来说在探究蛋白质构象变化方面,使用单一的CV(反应坐标),例如DISTANCE,ANGLE。DIHEDRAL等反应坐标,往往不能够反应构象变化的具体机制,所以这时候可能有人会说那么将多种集合变量,即反应坐标放在一起进行重要性采样(metadynamics,ABF),实际上随着集合变量的数量的增加,计算所需要的计算量也会增加,其次则会导致在模拟的过程中覆盖真实的反应坐标,即由我们自己选择的变量所定义出来的反应坐标只是模拟的反应坐标,并非真实反应或者机制过程中的真实反应坐标,因此定义合适的反应坐标CV往往可以实现在反应坐标空间内的全采样。
http://1.US sampling
关于伞形采样的概念在比较早的时候就已经提出了,在gromacs中就可以实现基本的伞形采样的功能(详见gromacs上的官网教程),可以通过编写自定义的mdp从而实现,此方法一般可以用于探究多肽或者配体与蛋白质本身的结合解离机制,对于窗口的数量的选择与合适的窗口选择对于模拟的准确性起到了至关重要的作用,对于伞形采样,早期的后处理方法一般为加权直方图分析法,即WHAM(现在gromacs中内置的用于伞形采样的后处理分析方法也还是WHAM),假设你在模拟的过程中设立了n个窗口,每一个窗口分别可以使用w进行表示,则WHAM方法则是通过求解各个窗口w总而来合并所定义的窗口,在后处理的过程中WHAM本身比较依赖于窗口的大小,并且暂时没有办法去选择较为普适的最优窗口大小,所以伞状积分(Umbrella integrate)方法得以提出,加速了伞形采样增强采样的计算收敛速度,同时后期还有人提出了MBAR方法,在这里就不详细 叙述了,MBAR主要是在误差计算方面有所改进,相比于WHAM计算结果的误差更小,个人比较推荐使用UI或者是MBAR方法。虽然有很多的方法可以对于伞形采样的结果进行改善,但是US本身的缺陷或者说是劣势为需要人工手动调节偏置势的高低。后期发展的AUS以及REUS则一定程度上改善了US方法本身的不足。对应的方法和实现软件使用gromacs即可。
2.MetaDynamics
关于MetaD,也算是一个老朋友了,MetaD为元动力学,当一个体系陷入自由能最低点或者是局部最小值点状态的时候,会导致通过cMD的方法在反应坐标上的采样不足问题,所以MetaD针对这样的情况则是在反应坐标上以一定的频率施加偏置势,这样就可以使整个体系进一步走出自由能的局部极小值点,从而推动整个模拟探索更广泛的化学空间,最后在通过施加的偏置势总和来计算反应沿着反应坐标的自由能变化,其中施加的偏置势在多数情况下为高斯峰的形式,因此有关于高斯峰的参数在一定程度上也决定了模拟正确程度与走向,因此比较难以控制。在较为早期的MtD方法中高低峰高(即HILLHIGHET)为固定的。MetaD中反应坐标的自由能可以通过如下的计算公式得到:
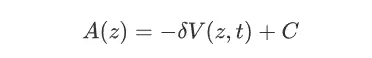
在这个函数中,V(z,t)为一个e为底的指数函数,所以当随着时间t的增长,V(z,t)并不会收敛到一个确定的数值,另外随着时间的增长计算模拟的复杂度也在不断上升,计算的速度也在不断变慢,因此为了解决这样的问题,后续有人提出了WTMetaD,在运用了well-tempered的方法之后可以有效的加快模拟过程中的收敛速度,还有MetaD结合了GRID,将偏置势离散化以降低了计算的复杂程度,目前位置,WTMetaD往往运用于蛋白质和配体之间的相互作用的情况比较多,当然也可以将MetaD与String-Method(弦理论)相互结合进一步更好的探究涉及到蛋白质构象变化的机制问题。关于可以实现metaD的软件主要有gromacs,NAMD,amber等等。
3.ABF method
接下来主要介绍一下文中的ABF方法,ABF与上述的两种经典方法都不太一样,ABF施加的是偏置力,而非偏置势,ABF属于先计算反应坐标上的平均力,然后施加并计算施加偏置力,最后在使用一定的积分方法计算得到相应的自由能,对于ABF实现较好的软件为NAMD以及其中的插件colvars,上述所谓的计算反应坐标的平均力可以映射到colvars配置文件中的fullsample,涉及到的积分计算则可以使用CZAR或者是较早版本的abf_integrator,与MetaD不同的是,ABF方法计算出来的梯度也会随着模拟时间的增加而收敛,所以ABF中的梯度也是衡量计算模拟结果是否已经收敛的重要方式之一,与此同时自由能的计算结果也会随着时间的增长而收敛,另外一个比较好的点,abf方法不需要像metaD一般设置需要猜测的参数如高斯峰的宽度和高度,下面以colvars中的两种方法的输入文件设置来作一下简单的阐述:
1.ABF
abf {
colvars psi d1 d2
fullSamples 1000
historyfreq 500000
writeCZARwindowFile on
CZARestimator on
integrate on
}2.MetaD
metadynamics {
colvars psi d1 d2
hillwidth 2.0
hillweight 0.1
welltempered on
biastemperature 4000
}有上述的简单代码其实可以看见,在MetaD中hillwidth以及hillweight都可能需要一定程度的猜测,同时这也给计算带来一定的复杂程度,如果设置的不合理则会带来一定的负面影响,一般都是在多次尝试下所得到的经验常数据多。如果想要在colvars中结合abf与metaD,或者说是使用metaD的方法,在colvars中定义相关的CV时,最好将expandboundaries选项开启从而使metaD的计算过程中覆盖所有的hill。上述的内容则是写在这篇文章之前的关于集中增强采样方法的简要介绍。
3.文章中的模拟方法和参数阐述
文章中所用到的蛋白质为1ELR,如下图所示:
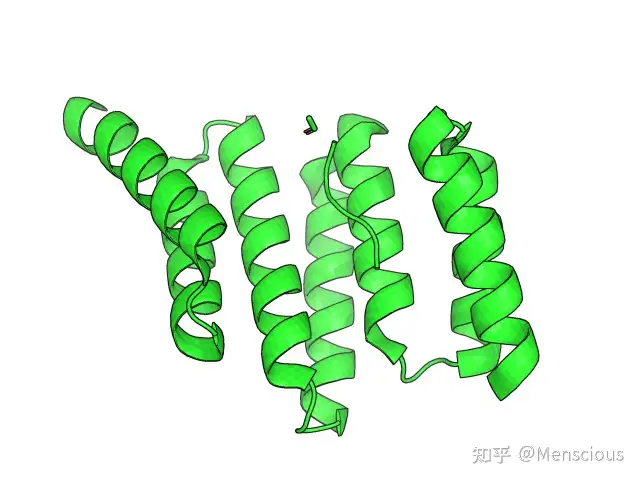
如图中所示,蛋白质中存在一条多肽,且多肽的C端Asp天冬氨酸和蛋白质之间形成了较为稳定的双羧酸夹持结构,为了探究多肽从蛋白质本身的解离机制,作者基本上按照如下的流程进行模拟。
TPR2A的初始结构与结合的肽段从PDB(PDB ID:1ELR)获取。蛋白质基质外部和内部的晶体水分子被移除。根据中性pH值,确定了离子化状态,组氨酸没有质子化,并添加了氢原子。建立了一个含有明确水分子的水盒来溶解蛋白质。该系统包括所有蛋白质原子和6230个溶剂中的水分子。使用NAMD v. 2.11和CHARMM36力场进行能量最小化和分子动力学模拟。模拟过程中的长程静电相互作用使用粒子网格Ewald(PME)求和计算,网格间距为1 Å。范德华相互作用在距离超过12 Å后被截断,共价键使用SHAKE算法固定。使用Langevin恒温器将温度控制在300 K,并且积分时间步长为2 fs。通过使用Langevin Nose−́ Hoover活塞(耦合常数为1 ps用于恒温器和0.5 ps用于压力计)在恒定压力(P = 1.01 bar)和温度(T = 300 K)下对系统进行了200 ps的平衡。使用VMD的Psfgen程序添加了氢原子,然后使用NAMD v. 2.11进行了10,000次能量最小化迭代。采用自适应偏置力计算(ABF)沿着一个被设定为距离r的集体变量(CV)进行。ABF基于累积力偏差,旨在抵消沿着CV的平均系统力,以加速穿越自由能势垒。ABF应用于受体-配体距离从8.5到30 Å的范围。CV距离分为两个窗口,一个窗口从8.5到20 Å,另一个窗口从20到30 Å。对于限制肽段相对于受体的角运动,应用了一个有平底的谐振势束缚。具体而言,由配体的一个原子(原子43,CA Glu 2)和受体的两个原子(原子358,CA Lys 238和原子1388,CA Lys 301)定义的角度θ被限制在可能从0到θ0 = 60°的振动范围内,使用带有上墙和下墙的谐振势和一个力常数为0.5 kcal/mol。这个角度采样限制连同下面的PMF径向积分限制定义了本工作中复合物的结合态。在每个窗口中进行了400 ns的ABF模拟,并且将瞬时力收集到0.1 Å的bin中。通过对ABF计算产生的平均力进行积分,得到了沿着受体-配体距离的平均力势(PMF),w(r)。在应用ABF偏置力之前,收集了2000个力样本。误差使用Comer等人在之前的工作中提出的公式进行计算。上述所设置的模拟设置可供参考也提供了一个非常好的示例。
